Deutschlandstipendium

Deutschlandstipendium an der TU Darmstadt
Aus Halbe-Halbe wird ein Ganzes
Das Deutschlandstipendium fördert besonders talentierte und engagierte Studierende und unterstützt damit die exzellente Ausbildung zukünftiger Fach- und Führungskräfte. Studierende erhalten 300 Euro monatlich: Die eine Hälfte der monatlichen Zuwendung kommt vom Bund und die andere Hälfte von Unternehmen, Stiftungen und privaten Förderinnen und Förderern. Die Übergabe der Stipendien erfolgt Jährlich im Januar im Rahmen einer feierlichen Zeremonie, unserer Stipendienfeier.
Wie es funktioniert
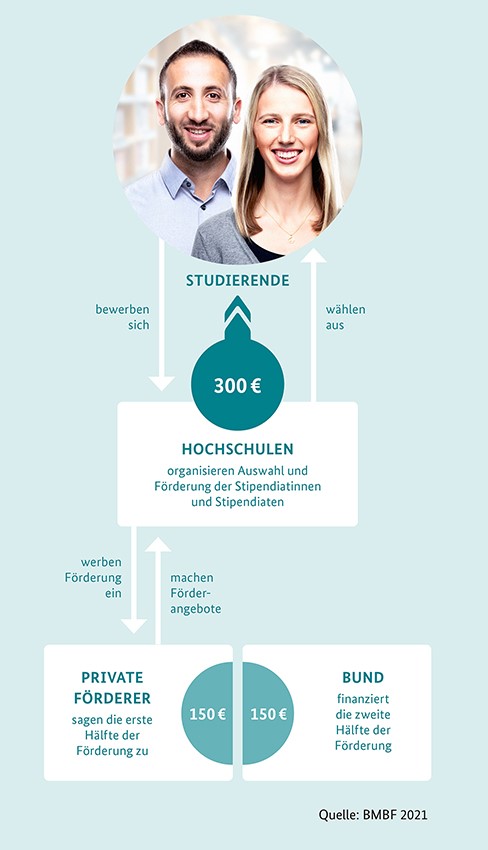

Über das Deutschlandstipendium werden seit dem Sommersemester 2011 Studierende sowie Studienanfängerinnen und Studienanfänger gefördert, deren Werdegang herausragende Leistungen in Studium und Beruf erwarten lässt. Sie erhalten 300 Euro monatlich – die eine Hälfte vom Bund und die andere Hälfte von privaten Stiftern. Dieses Bündnis aus zivilgesellschaftlichem Engagement und staatlicher Förderung ist das Besondere am Deutschlandstipendium.
Die Stipendiatinnen und Stipendiaten erhalten das einkommensunabhängige Fördergeld von monatlich 300 Euro für mindestens zwei Semester und höchstens bis zum Ende ihrer Regelstudienzeit. So können sie sich erfolgreich auf ihre Hochschulausbildung konzentrieren.
Erfolgreiche Partnerschaften für ausgezeichnete Talente
Die TU Darmstadt ist deutschlandweit eine der erfolgreichsten Hochschulen in der Einwerbung von Stipendien. Dies gelingt durch ein besonderes Matching-Verfahren , welches die Interessen der Studierenden und der Förderer bestmöglich zusammen bringt – beste Voraussetzung für erfolgreiche Partnerschaften!






